Persons
30167 Hannover
30167 Hannover
30167 Hannover
30167 Hannover
Research
Three good reasons why the response of atoms and molecules to strong light pulses is a subject of ongoing research are the following:
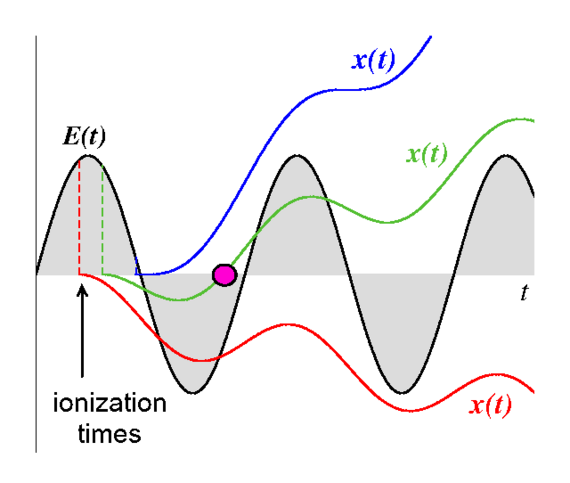

i- The laser-induced dynamics follows and reveals the laws of quantum mechanics with explicitly time dependent Hamiltonian in a regime where low-order time-dependent perturbation theory is not applicable. Strong-field dynamics in combination with Coulomb-interacting particles remains a challenge for theoretical study and often requires substantial numerical effort. At the same time, laser-driven atoms can show behaviour that is reasonably but not accurately enough explained by classical mechanics leading into the interesting regime where classical and quantum physics meet. Electrons that leave their parent ion after ionization by a field with low frequency and high field strength fall into this category.
ii- The ability to shape the incident field by adjusting a multitude of parameters such as intensity, wavelength, pulse duration, carrier-envelope phase, polarization, spectral composition and pulse shape allows us to control microscopic dynamics on ultrafast time scales. Ultimate goals of quantum control are the laser-based steering of chemical reactions and the controlled emission of charged particles and coherent radiation.
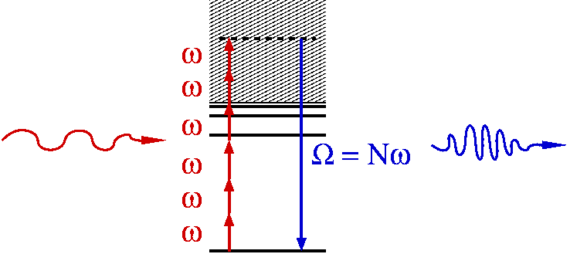
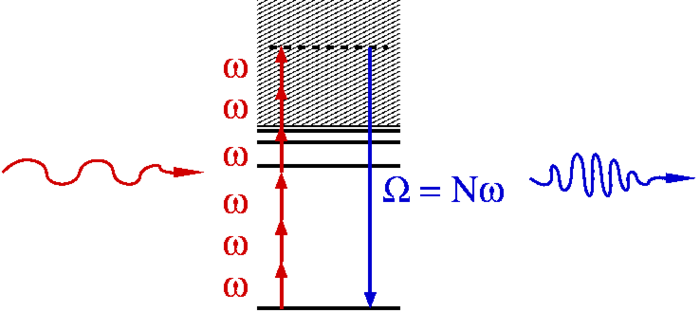
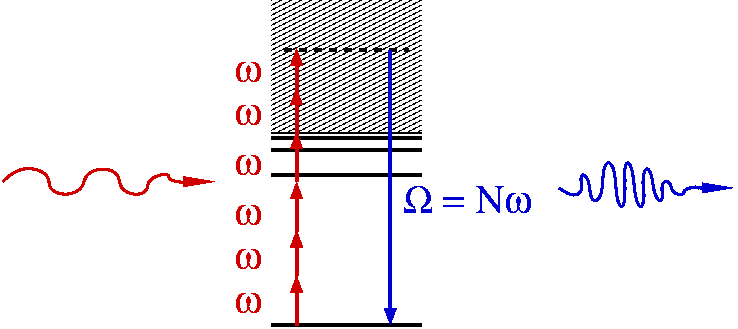
iii- Laser-irradiated atoms and molecules serve as a source of coherent high-frequency radiation with photon energies reaching from the UV into the X-ray range. This conversion process is known as high-harmonic generation and provides the basis for the formation of attosecond pulses, i.e. coherent pulses with durations below one femtosecond or even below 100 attoseconds. We know also that harmonic generation is due to laser-driven electrons that spend a sub-laser-cycle excursion time in an unbound state before returning into their initial bound state. The dependence of the excursion time on the photon energy offers the possibility to read attosecond-scale dynamics of the parent ion from the observed radiation.
Literature for ruther reading
- M. Kitzler and S. Gräfe (Eds.), Ultrafast Dynamics Driven by Intense Light Pulses
- C. J. Joachain, N. J. Kylstra and R. M. Potvliege, Atoms in Intense Laser Fields
- M. Shapiro and P. Brumer, Quantum Control of Molecular Processes
- P. Mulser and D. Bauer, High Power Laser-Matter Interaction
- F. Krausz and M. Ivanov, Attosecond physics





